Introduction
Humans are mediating the global dispersal of species at unprecedented rates, and the resulting biological invasions can have rapid, substantial effects on native species (
Ricciardi 2007;
Ricciardi et al. 2013). Contact among previously isolated species is a major driver of emerging infectious diseases in humans, plants, and animals, which threaten public health, food security, and biodiversity (
Fisher et al. 2012;
Voyles et al. 2015). Conservation genomics and conservation biogeography represent key tools in the fight against emerging diseases.
Neutral genetic structure of endangered host species informs the delineation of meaningful management units for threat mitigation. Genetic structure can sometimes predict the spread of host-dispersed pathogens by estimating the magnitude and direction of dispersal among populations, or identifying barriers to dispersal that could limit pathogen spread (
Blanchong et al. 2008;
Biek and Real 2010;
Wilder et al. 2015). Delimiting neutral population structure is also a prerequisite for assessing local adaptation because it allows the effects of genetic drift and selection to be separated (
Ekblom et al. 2007;
Spurgin and Richardson 2010;
Rico et al. 2016).
Functional genetic markers further inform the management of endangered species by identifying populations with strong local adaptation. In populations under pressure from virulent pathogens, immunogenetic local adaptation may be particularly relevant. Pathogens exert powerful selective pressures on naïve host populations (
Sironi et al. 2015), and genetic signatures of infection-related selective sweeps may be detectable for generations (
de Groot et al. 2002;
Di Gaspero et al. 2012;
Deschamps et al. 2016). Detection of selective sweeps on particular genes or gene families can confirm or exclude potential mechanisms of susceptibility (in the host) or virulence (in the pathogen), informing the development of treatments (
Campbell and Tishkoff 2008).
The recently emerged fungal pathogen
Pseudogymnoascus destructans causes white-nose syndrome (WNS) in hibernating bats (
Cryan et al. 2013). The fungus is endemic to Eurasia (
Leopardi et al. 2015) and was introduced to North America, where it was first observed in New York State in 2006 (
Blehert et al. 2009). Since 2006,
P. destructans has spread rapidly, causing local extinctions and widespread population declines in some Nearctic species of bat, including the little brown myotis (
Myotis lucifugus LeConte, 1831), northern myotis (
Myotis septentrionalis), and Indiana myotis (
Myotis sodalis) (
Langwig et al. 2012;
Frick et al. 2015). Bats are exposed when they enter hibernacula containing
P. destructans. Fungal lesions may develop on the wings during hibernation and disrupt homeostasis, triggering a cascade of behavioural and physiological responses that result in high mortality rates in susceptible species of bats (reviewed in
Willis 2015).
The spread of WNS is associated in part with genetic structure among populations of
M. lucifugus (
Miller-Butterworth et al. 2014;
Davy et al. 2015;
Wilder et al. 2015;
Vonhof et al. 2016). Genetic, ecological, and environmental data have been used to model the direction and speed with which bats may facilitate dispersal of
P. destructans towards the Pacific Coast of North America (
Maher et al. 2012;
O’Regan et al. 2015). However, models cannot fully predict pathogen dispersal, as illustrated by the recent, unexpected emergence of WNS in western Washington State in March 2016 (
Lorch et al. 2016). Previous, large-scale population genetics studies (
Vonhof et al. 2015;
Wilder et al. 2015) suggest that genetic structure in
M. lucifugus may be higher in the western portion of its range. If these patterns can be clarified with more targeted sampling, they may predict the path along which
P. destructans is most likely to spread from its new “western front”.
Tolerance of WNS in Eurasian bats is attributed to coevolution with
P. destructans (
Zukal et al. 2016), and a similar evolutionary process may currently be underway in North American bats (
Frick et al. 2017). In four years, following the initial, precipitous declines from WNS, estimated annual adult survivorship of
M. lucifugus in some populations increased from 0.68 to 0.75 (for males), and from 0.65 to 0.70 (for females;
Maslo et al. 2015). Local immunogenetic adaptation to
P. destructans provides one potential explanation for this trend, and the ubiquity of
M. lucifugus and its transcontinental range allow extensive sampling across a large geographic area. Thus,
M. lucifugus provides an excellent model for investigating local immunogenetic adaptation in bats, and ultimately for testing whether WNS exerts selection on immune genes.
Following the recent western introduction of
P. destructans (
Lorch et al. 2016), management biologists need detailed data on directional dispersal in
M. lucifugus in northwestern North America to prioritize areas for surveillance and mitigation efforts. The range-wide approach of previous studies (
Vonhof et al. 2015;
Wilder et al. 2015) provides a valuable perspective on the contemporary genetic structure of this endangered species, but small sample sizes from northwestern locations (Manitoba to British Colombia, and northward to Alaska) limit their resolution. Here, we investigated fine-scale contemporary neutral genetic population structure in 1142
M. lucifugus sampled across the species’ northern range. We tested for genetic population structure and used Bayesian estimates of gene flow to explore the most likely direction of spread from the new source in Washington State. We also estimated the effective population size for each identified population. Finally, we used 454 pyrosequencing of the DRB1-like exon 2 of the MHC to explore immunogenetic adaptation of
M. lucifugus to
P. destructans. We hypothesized that populations of
M. lucifugus sampled prior to the arrival of WNS were locally adapted to existing selective pressures on the MHC, predicting discordant patterns of structure between neutral and immunogenetic markers. Finally, we tested the hypothesis that WNS exerts direct selection on the MHC by comparing DRB1 diversity in eastern Canada before and after the arrival of WNS.
Discussion
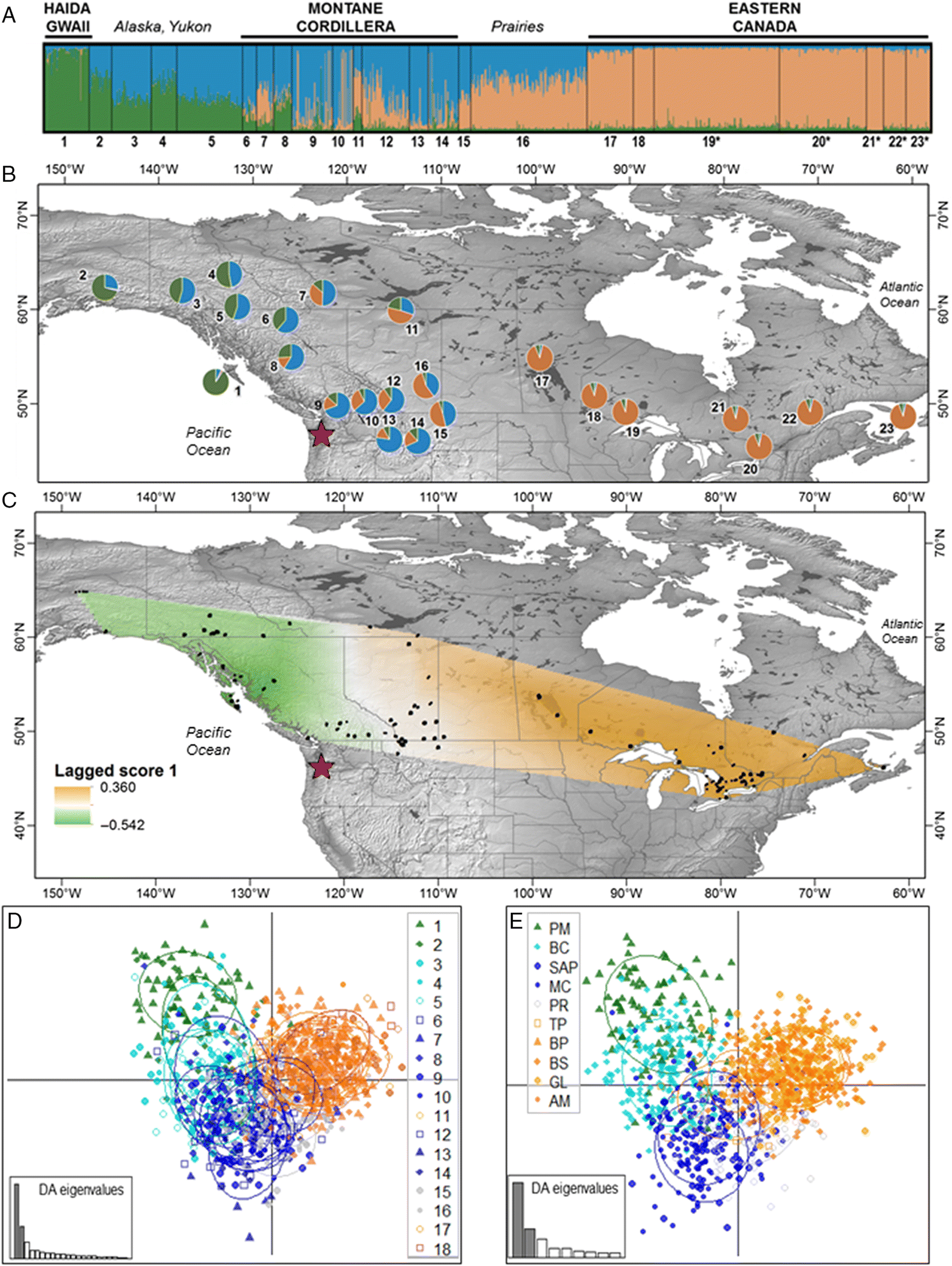
Neutral genetic structure in northern populations of
M. lucifugus generally increases from east to west, and the spatial distribution of neutral genetic diversity across the northwestern range of
M. lucifugus is intriguing: neither the Coastal nor Rocky Mountains act as barriers to gene flow, yet bats on Haida Gwaii appear to be effectively isolated. Our results predict that Haida Gwaii may act as a temporary refugium from the disease, whereas
M. lucifugus may carry
P. destructans more quickly through southern BC, across the Rockies, and into Montana and Alberta. Southward gene flow from the Yukon opposes the direction of the oncoming pathogen, providing some hope that bats in the Yukon and Alaska may experience the same delay in
P. destructans arrival that has occurred in central Canada (
Davy et al. 2015;
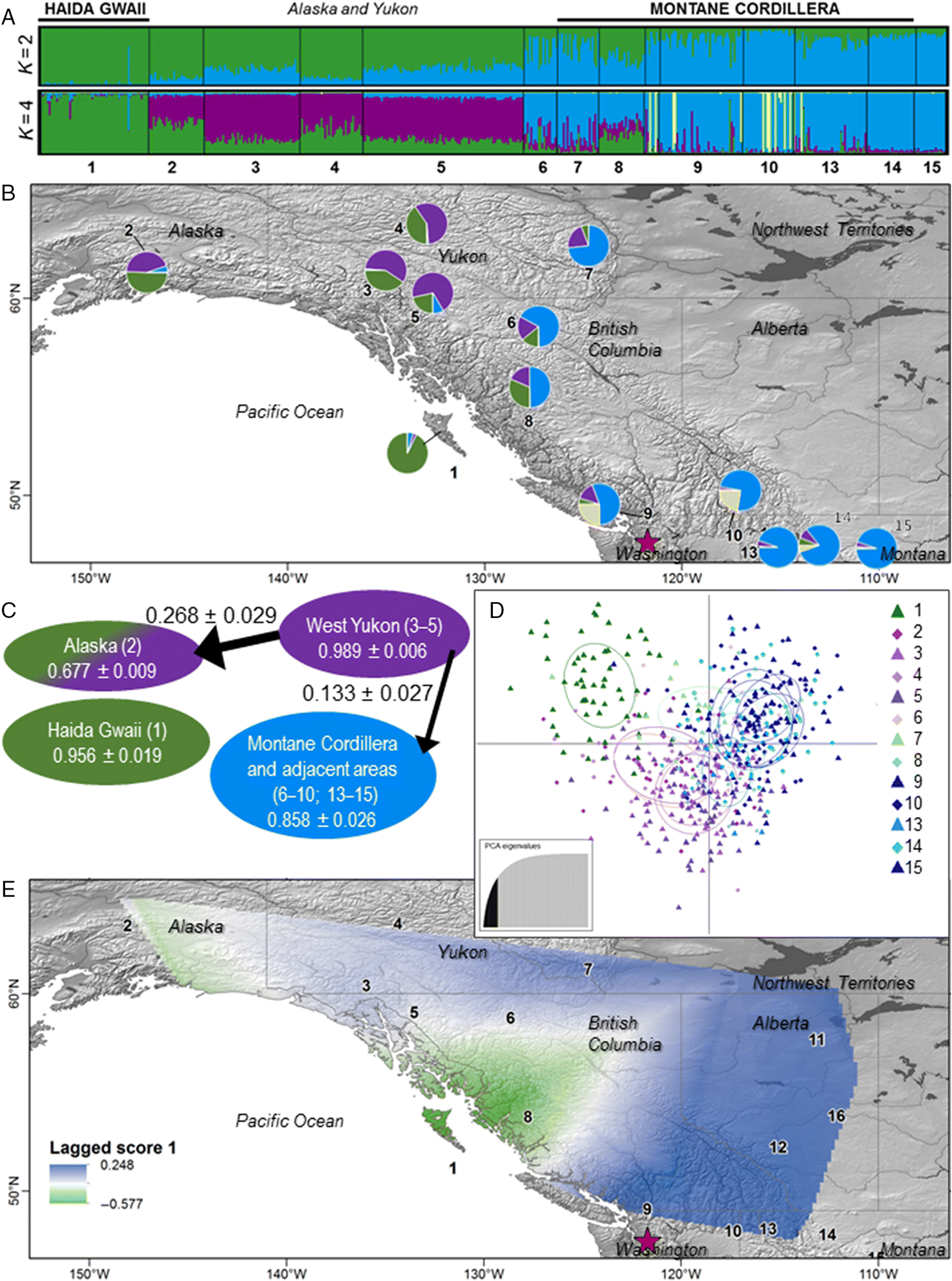
Wilder et al. 2015). We detected unexpected fine-scale population structure in the Yukon, with differentiation between the eastern and western Yukon sampling areas (
Figs. 2A,
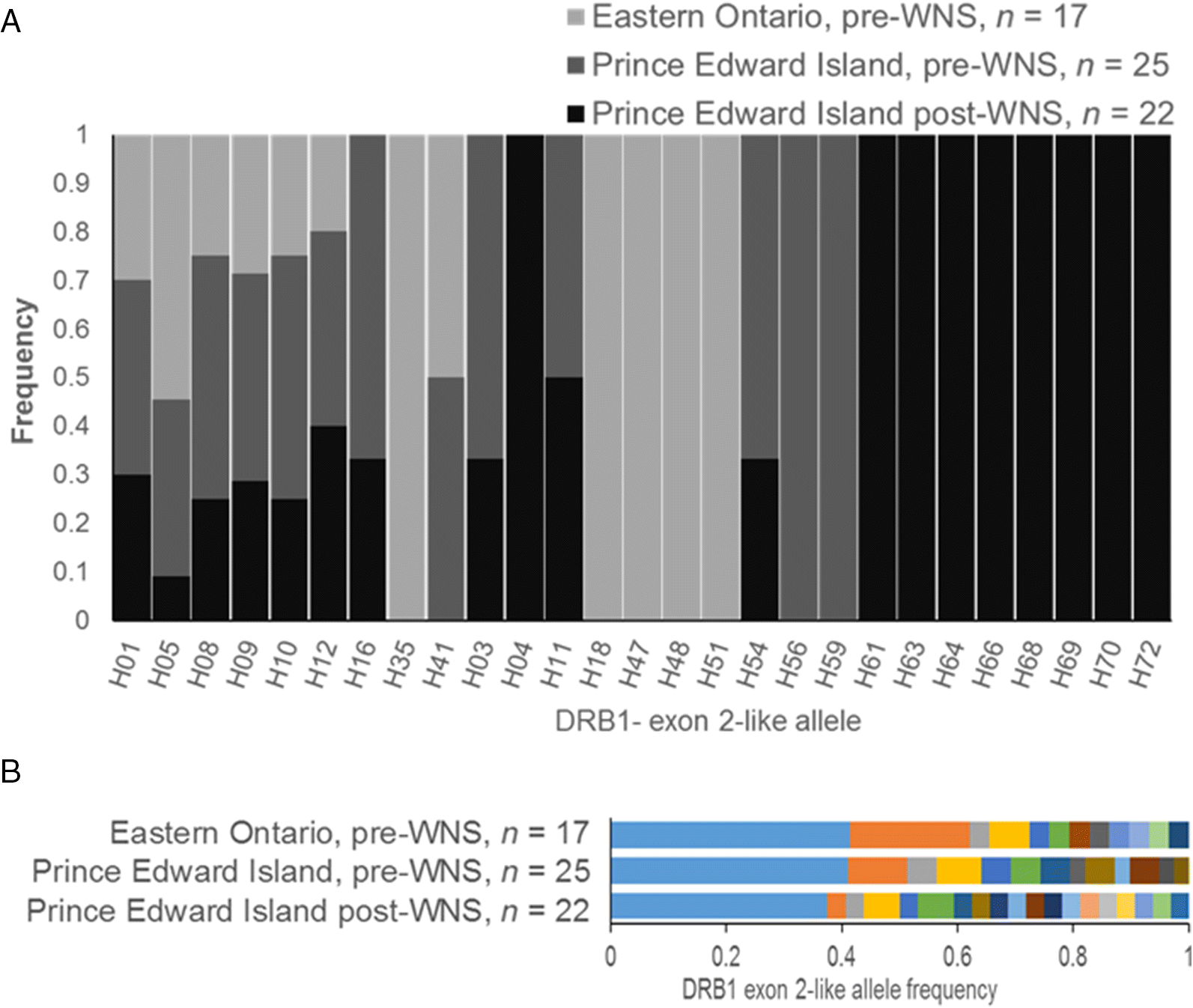
2B). Preliminary immunogenetic analysis suggests that baseline patterns of MHC may be altered by the impacts of WNS. This could indicate that the MHC is under balancing selection that is disrupted by the arrival of WNS. However, our immunogenetic analyses were confounded by high gene duplication in this species (see also
Palmer et al. (2016), who detected 24 DRB1-like alleles in only 15 individuals), and new sequencing methods will be required to disentangle the immunogenetic impacts of WNS on bats. We also relied on samples from bats that died in the first waves of mortality following WNS. Although our results are suggestive of selection by WNS on the MHC, we were not able to confirm or refute this hypothesis.
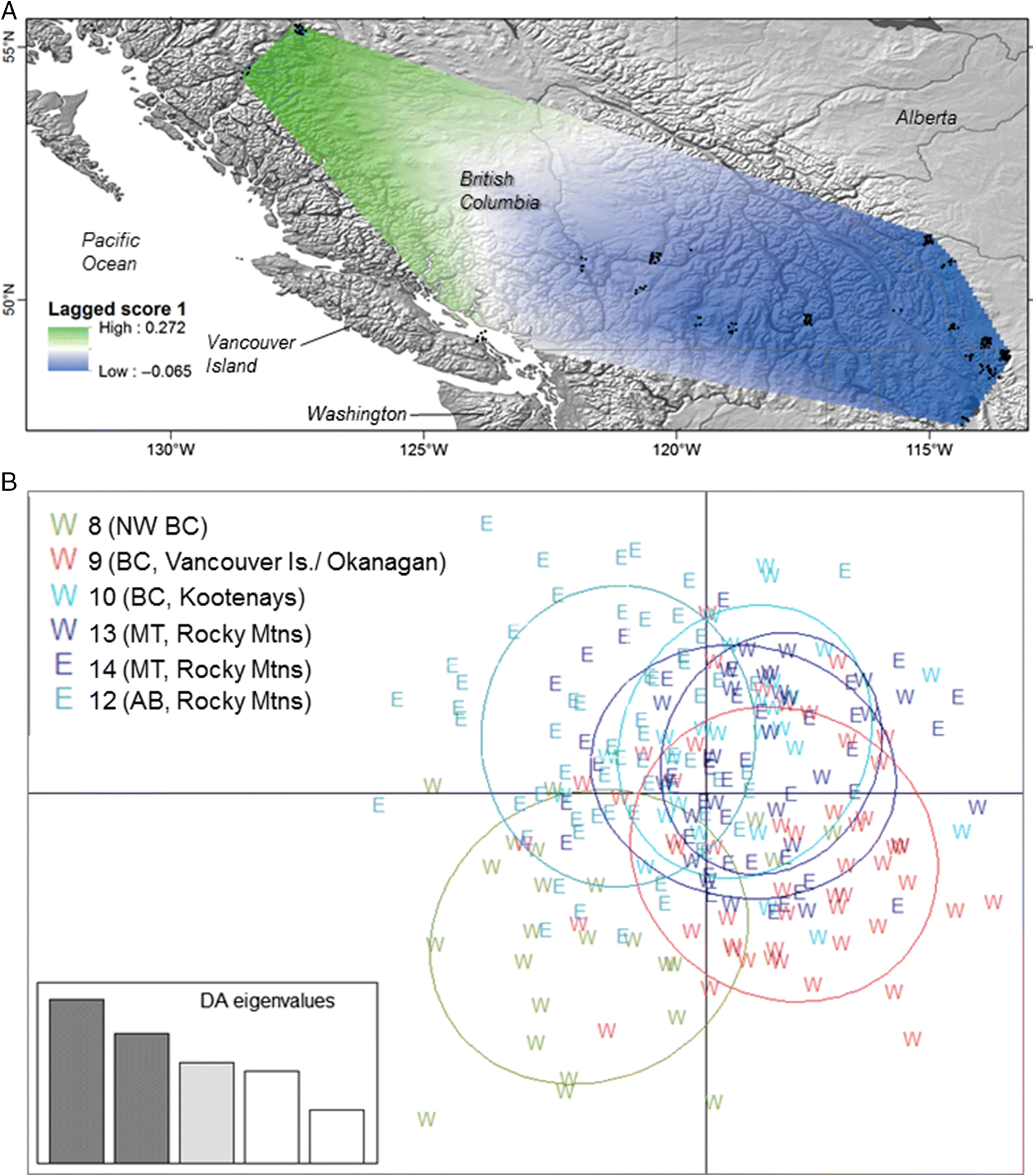
Estimates of effective population size are surprisingly low given
M. lucifugus’ ubiquitous distribution and high genetic diversity, which are often associated with extremely high
Ne. Our estimates were 10–1000× lower than estimates for three migratory bat species (
Lasionycteris noctivagans,
Lasiurus cinereus, and
Lasiurus borealis), based on samples from a single wind farm (
Sovic et al. 2016). Estimates of
Ne based on single samples can be affected by many factors, and the values presented here should not be misinterpreted as an estimate of the absolute number of breeding individuals. Rather, the estimates indicate relative effective population sizes among genetic clusters. The low values estimated here suggest that despite its historical ubiquity,
M. lucifugus may be unexpectedly vulnerable to population declines from additive mortality, with more reproductive variation among individuals than previously thought. The particularly low
Ne of western populations may leave them more vulnerable to declines once the pathogen arrives, although the genetic structure of
M. lucifugus in the west may slow the spread of WNS.
Our results expand on previous studies of
M. lucifugus (
Burns et al. 2014;
Davy et al. 2015;
Vonhof et al. 2015;
Wilder et al. 2015) by clarifying the locations of genetic transitions among western bat populations, where previous studies already detected increased genetic variation, and revealing further population structure in the west, particularly on Haida Gwaii. Haida Gwaii bats apparently exhibit low dispersal across the Hecate Strait, and dispersal towards the island is negligible. Haida Gwaii acted as a glacial refugium during the Quaternary glaciation, as did parts of Alaska and the Yukon, and the genetic endemism of bats on Haida Gwaii mirrors that found in other taxa, including a sea star (
Patiria miniata), the black bear (
Ursus americanus), and several land birds (
Byun et al. 1997;
Keever et al. 2009;
Pruett et al. 2013). Although
M. lucifugus undertake migrations >500 km (
Fenton 1969;
Norquay et al. 2013) and disperse to and from Vancouver Island, Newfoundland, and PEI (
Burns et al. 2014; this study), our results suggest that the longer flight distance to Haida Gwaii from adjacent coastal islands (≥48 km) and the prevailing westerly winds may deter bats from crossing. Nevertheless, accidental translocation of bats on boats could move individuals across the Hecate Strait (e.g.,
Constantine 2003;
Wright and Moran 2010). We strongly recommend that stringent preventative measures and public outreach are used to avoid human dispersal of
P. destructans to the archipelago and delay the pathogen’s arrival for as long as possible.
Previous studies suggested that mountain ranges may limit population connectivity in
M. lucifugus (
Vonhof et al. 2015;
Wilder et al. 2015), but our sampling design revealed that the mountain ranges are not barriers to dispersal in this species. In our study area, the closely related
M. californicus and
M. yumanensis occur only on the western side of the Continental Divide (C.L. Lausen, unpublished data), indicating that the mountains act as a barrier. In contrast, genetic clusters of
M. lucifugus align with ecozones, suggesting possible “montane”, “prairie”, and “coastal” ecotypes. Morphological differences in
M. lucifugus including pinnae length and forearm length support this hypothesis, also corresponding to ecozone boundaries (
Lausen et al. 2008; C.L. Lausen and T.S. Jung, unpublished data). Future research can test potential behavioural or morphological differences among putative ecotypes that could explain their continued differentiation such as differences in movement, communication, hibernation, or foraging behaviour (
Veselka et al. 2013;
Jung et al. 2014;
Pond et al. 2016).
In the western Yukon,
M. lucifugus take advantage of insect-rich summer feeding grounds, but apparently migrate over the Coast Mountains to hibernate in talus slopes and tree-root wads of coastal Alaska, where winters are milder (
Jung et al. 2014;
Blejwas et al. 2015). Movement of Yukon bats into Alaska is also supported by our data, and by anecdotal reports of bats frozen into glaciers of the Coast Mountains (
Slough and Jung 2008). In the eastern Yukon, karst formations provide potential hibernacula and remove the need for energetically costly winter migrations. Thus, different availability of overwintering sites may drive the observed differentiation between bats in western and eastern Yukon. As above, morphological data (forearm length) also differentiate bats from the western and eastern Yukon (
Lausen et al. 2008;
Talerico 2008; C.L. Lausen et al., unpublished data).
The Canadian prairies appear to represent a zone of admixture between
M. lucifugus from the Eastern and Montane Cordillera clusters; a pattern that may be maintained by annual migrations among maternity, swarming, and overwintering sites, or that may represent the presence of a further, distinct genetic cluster that was not detected by our analyses. Maternity colonies of
M. lucifugus occur in anthropogenic structures on the Great Plains during the summer. Our results suggest that many of the
M. lucifugus using prairie maternity colonies must migrate to the edges to swarm (mate) and hibernate, restricting genetic exchange to the periphery of the prairies and preventing differentiation of the prairie population. No
M. lucifugus hibernacula are known in Saskatchewan or from the Alberta prairies, although hibernacula are known to the north in the Alberta boreal (
Reimer et al. 2014; C.L. Lausen et al., unpublished data) and to the west in the Alberta Rocky Mountains (
Schowalter 1980). Collecting genetic samples from these sites in future may provide further resolution on the ancestry of prairie bats and their relationship with surrounding genetic clusters.
Despite genetic population structure and high MHC polymorphism,
M. lucifugus exhibit low spatial immunogenetic structure across the northern extent of their range. The lack of MHC differentiation observed despite low levels of gene flow may reflect balancing selection prior to the introduction of WNS, from immunogenetic selective pressures on
M. lucifugus that may have been relatively constant among locations. However, gene duplication at the DRB1 locus limits the use of this data set. Specific MHC alleles have been linked to disease susceptibility in other host-pathogen systems, including amphibian resistance to the pathogenic fungus
Batrachochytridium dendrobatidis and the susceptibility of giant pandas (
Ailuropoda melanoleuca) to intestinal parasitic infections (
Savage and Zamudio 2011;
Bataille et al. 2015;
Zhang et al. 2015). Sequencing approaches that can distinguish between alleles from duplicated genes are required to further test the hypothesis that WNS exerts immunogenetic selection.
The idea that bats in western North America may harbour greater potential for the evolution of disease resistance based on standing genetic variation (
Wilder et al. 2015) is extremely attractive, especially given our current inability to prevent or cure WNS. The link between standing genetic variation within a population and adaptive potential with regard to shifting selective pressures is a central tenet of conservation genomics, but unfortunately, it does not always apply in practice (
Rollins et al. 2013;
Mittell et al. 2015). Bat WNS provides an excellent illustration of high standing genetic variation providing little protection in the face of emerging infectious diseases.
Myotis lucifugus was one of the most common, widespread and abundant mammal species in North America prior to the arrival of WNS. Heterozygosity of neutral markers and allelic diversity of at least one functional marker are high across the species’ range. Nevertheless, an emerging pathogen has brought this species to the brink of regional extinction in less than a decade. The new selective pressure to which northeastern populations of
M. lucifugus were exposed (i.e., WNS) is simply not one to which those populations could quickly adapt; at least not without high individual mortality rates. Although the conservation of genetic diversity is critical to long-term species conservation, it is not a guarantee of adaptive potential. Our preliminary results also indicate that although
M. lucifugus in western North America exhibit neutral genetic differences compared with the eastern population, this is not reflected in a functional gene expected to experience direct selection from WNS.
Selective sweeps from emerging diseases such as WNS could erode immunogenetic adaptations to other preexisting or future pathogens. The lack of regional MHC differentiation in
M. lucifugus across its northern range suggests that disease-related selective pressures on this species may be similar among genetically differentiated clusters, but testing this hypothesis requires more robust genetic tools that can differentiate among duplicated loci, and that can simultaneously target multiple immune genes. Bats in our sampling area coexist with a variety of pathogens including several strains of bat rabies (
Middleton et al. 2015), corona and polyoma viruses (
Misra et al. 2009), endemic fungal pathogens such as
Trichophyton redelli (
Lorch et al. 2015), internal parasites such as
Trypanosoma myoti (
Bower and Woo 1981), and ectoparasites such as fleas and mites (
Webber et al. 2015). Most of these are relatively widespread, affecting bats across North America. If WNS causes a significant selective sweep at MHC in
M. lucifugus across the continent, bats that survive WNS may experience a trade-off in susceptibility to other pathogens; a carryover effect of the disease that extends its influence past the stage of acute infection and recovery. Examples of other potential carryover effects from WNS include delayed parturition (
Francl et al. 2012) and increased chronic stress following recovery (
Davy et al. 2017).
Emerging infectious diseases provide excellent opportunities to document selective sweeps in real time, and to test hypotheses about the impacts of disease on genetic and phenotypic diversity. This requires a backdrop of robust data on neutral genetic diversity against which immunogenetic diversity can be contrasted (
Ekblom et al. 2007;
Rico et al. 2016). However, studies of immune response to infectious diseases rarely consider more than one pathogen at a time and often focus on particular immune genes (as we have done here). In reality, the immune system responds to a myriad of pathogens and other stressors simultaneously, and no free-ranging population is likely to coevolve in isolation with a single pathogen. The impacts of emerging infectious diseases on immunogenetic variation should, therefore, be considered in the context in which they emerge—as novel selective pressures on host populations that already have long evolutionary histories with a complex set of other pathogens, parasites, and commensal microflora.