Relaxin peptides reduce cellular damage in cultured brain slices exposed to transient oxygen–glucose deprivation: an effect mediated by nitric oxide
Abstract
The effect of treatment with human relaxins on cell death was studied in oxygen- and glucose-deprived brain slices. In addition, involvement of nitric oxide and the relaxin receptor, RXFP3, was studied. Brain slices (n = 12–18/group) were cultured under standard conditions for two weeks and then exposed to: (i) an oxygenated balanced salt solution, (ii) a deoxygenated, glucose-free balanced salt solution (OGD media), or (iii) OGD media containing 10−7 mol/L H2 relaxin, 10−7 mol/L H2 relaxin with 50 μmol/L L-NIL, 10−7 mol/L H3 relaxin, or 10−7 mol/L H3 relaxin with 50 μmol/L L-NIL. Cell death was assessed using propidium iodide fluorescence. In a separate experiment, 10−5 mol/L R3 B1-22R (an antagonist of RXFP3) was added to both H2 and H3 relaxin treatments. H2 and H3 relaxin treatment reduced cell damage or death in OGD slices and L-NIL partially attenuated the effect of H3 relaxin. Antagonism of RXFP3 blocked the effect of H3 but not H2 relaxin. These data increase our understanding of the role of relaxin ligands and their receptors in protecting tissues throughout the body from ischemia and reperfusion injury.
Introduction
Phylogenetic analyses suggest that relaxin ligand and receptor genes originated from three loci in ancestral vertebrates: ancrln giving rise to all relaxins and ancrxfp1/2 and ancrxfp3/4 giving rise to relaxin family peptide receptors (RXFPs) (Yegorov et al. 2014). In humans, three relaxins (H1, H2, and H3) and 4 insulin-like peptides (3, 4, 5, and 6), along with insulin comprise the insulin peptide superfamily. Four relaxin receptors have been identified, two with leucine-rich ectodomains and a LDLa module are classed as two type C GPCRs within Family A: RXFP1 and RXFP2. Two receptors, RXFP3 and 4 lack this ectodomain and are smaller than RXFP1 and 3 (Bathgate et al. 2013, 2018). The main circulating relaxin in humans (H2) is homologous to relaxin-1 in other mammals and by convention, is often called just “relaxin” (Bathgate et al. 2013). In addition, all mammals studied to date possess a relaxin neuropeptide, called relaxin-3 (H3 relaxin in humans), which is highly expressed in the dorsal tegmentum with immunoreactive fibres running forward to target receptor-rich zones in the forebrain (Bathgate et al. 2002; Burazin et al. 2002; Smith et al. 2010, 2011). Its amino acid sequence is highly conserved amongst vertebrates. In the forebrain, relaxin-3 modulates neuroendocrine responses to stress (Banerjee et al. 2010; Smith et al. 2010; Watanabe et al. 2011), induces feeding and drinking behaviours (McGowan et al. 2005, 2006, 2007; Sutton et al. 2009), and stimulates gonadotropin release (McGowan et al. 2008). RXFP1 is believed to be the cognate receptor for relaxin, whereas RXFP3 is believed to be the relaxin-3 receptor (Bathgate et al. 2013). However, receptor promiscuity has been demonstrated in vitro with H2 relaxin binding RXFP1–3 and H3 relaxin binding RXFP1, 3, and 4 (Liu et al. 2003a, 2003b; Sudo et al. 2003; Halls et al. 2007; van der Westhuizen et al. 2010). In cell culture, both H2 and H3 relaxin activate these receptors to varying degrees.
Recent reports have indicated that relaxin may protect tissues in organs such as the lungs (Alexiou et al. 2010, 2013), pancreas (Cosen-Binker et al. 2006), intestines (Masini et al. 2006; Matheson et al. 2014), kidney (Collino et al. 2013a, 2013b; Yoshida et al. 2013, 2014), liver (Boehnert et al. 2008; Kageyama et al. 2018), and heart (Masini et al. 1996, 1997; Bani et al. 1998; Dschietzig et al. 2001; Boccalini et al. 2015; Valle Raleigh et al. 2017; Beiert et al. 2018; Martin et al. 2018) from the detrimental effects of ischemia. The protective effects of relaxin are numerous and include vasodilation (Bani et al. 1998; Dschietzig et al. 2001; Failli et al. 2002; Nistri and Bani 2005), suppressing platelet aggregation (Bani et al. 1995), reducing mast cell granule release (Masini et al. 1994; Bani et al. 1998; Nistri et al. 2008), reducing neutrophil migration (Bani et al. 1998) and activation (Masini et al. 2004), and protecting cells from apoptosis (Moore et al. 2007). In two rodent models of stroke, a permanent middle cerebral artery occlusion (MCAO) model and a MCAO model where blood flow is restored permitting reperfusion, intravenous and intracerebral H2 and H3 relaxin significantly reduced the size of the resultant infarcts in the somatosensory cortex 4 h post-MCAO (Wilson et al. 2006; Bergeron et al. 2015). The response to cerebral ischemia is complex, thus there are numerous mechanisms by which relaxin may be eliciting tissue protective effects. However, many of these appear to be nitric oxide (NO)-mediated (Masini et al. 1995; Cosen-Binker et al. 2006; Wilson et al. 2006), and in the MCAO studies discussed above, treatment with L-NIO, a nonspecific inhibitor of NO synthase (NOS), blocked the effect of both relaxins on infarct development in MCAO rats (Bergeron et al. 2015).
The objectives of the current study were to determine if treatment with H2 or H3 relaxin reduced cellular damage and death in organotypic brain slice cultures deprived of both oxygen and glucose (OGD) for 1 h and also to elucidate the role of RXFP3 and NOS II in any observed protective effects.
Materials and methods
Animals
Sprague-Dawley rats (Charles River Laboratories, St. Constant, QC) were used in these experiments. All experimental procedures were carried out in accordance with guidelines established by the Canadian Council on Animal Care (CCAC 1993) and were approved by the Animal Care Committee at Acadia University, Wolfville, NS, Canada (protocol 10–18). Day 10–12 neonatal rat pups were used in organotypic brain slice culture experiments (methods derived from Strasser and Fischer (1995) and Gahwiler et al. (1997, 2001)). Timed pregnant dams were shipped to arrive at day 15 of pregnancy. Dams were resident in the Weston Animal Care Centre for approximately 1 week prior to term.
Organotypic brain slice cultures
Pups from 6 litters were randomly assigned to treatment groups. On days 10–12 postpartum, pups were euthanized by cervical dislocation and brains were removed for slicing. A 1-cm3 section of brain tissue, containing the hypothalamus and sensory cortex, was secured to the cutting stage of a fresh tissue vibratome (Vibroslice; Campden Instrument Company, Lafayette, IN, USA) using cyanoacrylate glue. This stage was placed into a reservoir containing chilled dissection media (500 mL: 1 × Minimum Essential Media with 0.6 g Tris, pH 7.2, filtered, bubbled with 95% O2/5% CO2) such that the brain tissue was fully immersed. Coronal slices (400 μm) were cut at the level of the commissural part of the anterior commissure where the columns of the fornix were visible (Paxinos and Watson 1998). At this level, the slices contained hypothalamus and sensory cortex. Under aseptic conditions in a laminar flow hood, two slices per brain were placed onto tissue culture inserts (MilliCell CM culture plate inserts; Sigma Aldrich, St. Louis, MO, USA) using natural hair paintbrushes. Inserts were then placed into wells of a 6-well culture plate (Corning Costar; Sigma Aldrich, St. Louis, MO, USA) with 1.1 mL of oxygenated culture media (500 mL: 250 mL 1 × Minimum Essential Media with 4 mmol/L L-glutamine, 12.95 mL HEPES, 125 mL 10 × Hank’s Balanced Salt Solution, 125 mL heat-inactivated horse serum, 37.5 g NaHCO3, 5 mL antibiotic/antimycotic and 0.3 g Tris; Sigma Aldrich, St. Louis, MO, USA). Plates were incubated for two weeks at 37 °C in 5% CO2/balance air prior to experimentation. Media was changed under aseptic conditions at 24 h, and then every 48 h during the two weeks of incubation. At two weeks of culture, one brain slice from each plate was stained with 2% TTC (2,3,5-triphenyltetrazolium chloride, Sigma Aldrich, Oakville, ON) to test for viability.
Treatments
Effect of H2 and H3 relaxin on neural cell survival: role of NO
Under aseptic conditions, culture media was aspirated from each well and replaced with 1 mL of one of 6 treatments (n = 12–18 slices per group) for 60 min: (1) controls (balanced salt solution: 120 mmol/L NaCl, 5 mmol/L KCl, 1.25 mmol/L NaH2PO4, 2 mmol/L MgSO4, 2 mmol/L CaCl2, 25 mmol/L NaHCO3, 10 mmol/L D-glucose and 20 mmol/L HEPES; Sigma Aldrich, St. Louis, MO, USA: 37 °C, pH 7.2, oxygenated by bubbling with 95% O2/5%CO2 for 30 min), (2) oxygen- and glucose-deprived (OGD media: balanced salt solution as above with 10 mmol/L sucrose replacing D-glucose, de-oxygenated by bubbling with 5% CO2/95% N2 for 30 min), (3) OGD media with 100 nmol/L H2 relaxin (035-62, human recombinant H2 relaxin, Phoenix Pharmaceuticals Inc., Burlingame, CA), (4) OGD media with 100 nmol/L H2 relaxin and 50 μmol/L L-NIL (NOSII inhibitor, Caymen Chemical Company, Ann Arbor, MI, USA), (5) OGD media with 100 nmol/L H3 relaxin (035-36, human recombinant H3 relaxin, Phoenix Pharmaceuticals Inc.) or (6) OGD media with 100 nmol/L H3 relaxin and 50 μmol/L L-NIL. Culture plates were returned to the incubator for 60 min. A separate group of cultured slices were used as a baseline control for measures of propidium iodide positive (PI+ve) cells. Following two weeks of culture, these slices were not treated, but stained with PI and the number of dead cells counted as above.
Role of RXFP3
Following an identical protocol to that above, separate groups (n = 10–14) of cultured brain slices were treated with: (1) Control media, (2) OGD media, (3) OGD media with 100 nmol/L H2 relaxin, (4) OGD media with 100 nmol/L H2 relaxin and 10 μmol/L R3 B1-22R (RXFP3 antagonist, Howard Florey Institute, Melbourne (Haugaard-Kedstrom et al. 2011)), (5) OGD media with 100 nmol/L H3 relaxin, and (6) OGD media with H3 relaxin and 10 μmol/L R3 B1-22R. Once again, slices were returned to the incubator following the 60 min treatment period for an hour prior to PI staining.
Determination and analysis of cell viability
Following treatment, solutions were aspirated, replaced with new culture media (37 °C, oxygenated), and returned to the incubator for 1 h. Twenty min prior to imaging, 5 μg/mL of propidium iodide (Sigma Aldrich, St. Louis, MO, USA) was added to culture wells. PI will only stain cells with permeated plasma membranes, which can then be used to assess cell viability. Slices were placed in wet mounts on slides and digitally imaged with an epifluorescence microscope (Nikon Eclipse TE2000U; Nikon Instruments, Melville, NY, USA) at 40× power using a green filter. The fluorescence excitation maximum of propidium iodide is 535 nm and the emission maximum is 617 nm. In each brain slice, PI+ve cells were counted in a sample window (300 × 300 pixels) in the S1 cortical region (Paxinos and Watson 1998) using Image J (Version 1.48v, Image Processing and Analysis in Java, Betheseda, MD) by two independent people blind to treatment condition. Mean numbers of PI+ve cells were compared between groups using one-way ANOVA with Tukey’s posthoc mean comparison test using GraphPad Prism version 6.0a for Mac OS X (GraphPad Software, San Diego, CA, USA). Means were considered significantly different at p < 0.05.
Results
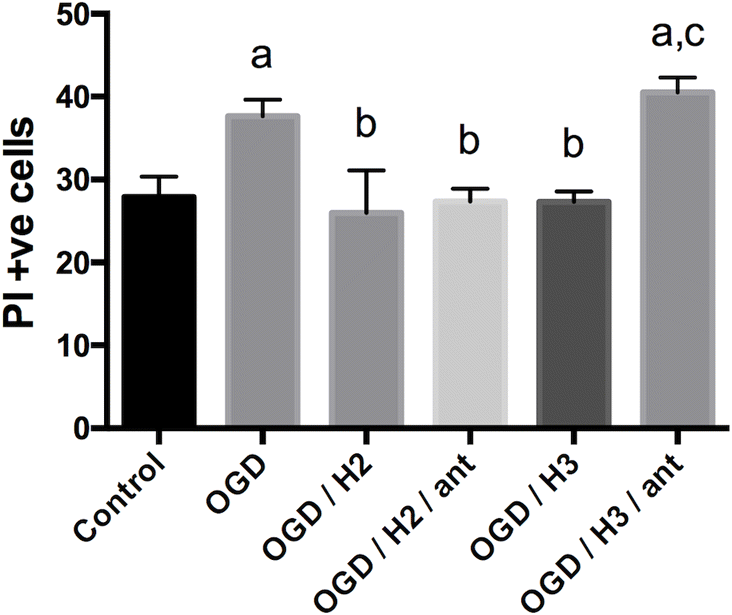
After 2 weeks in culture, TTC staining suggested that brain slices in each plate were viable. Fluorescent cell counting in a 300 × 300 pixel cortical sample window of PI stained slices (Fig. 1) showed an average of 20 dead or damaged cells (baseline; Fig. 2). The number of PI+ve cells in slices receiving a 60 min control treatment with oxygenated, balanced salt solution with glucose was not significantly different (p > 0.05; Fig. 2) from baseline. In both experiments, oxygen and glucose deprivation significantly (p < 0.05) increased cell damage or death compared with controls (Figs. 2 and 3), and treatment with 100 nmol/L H2 or H3 relaxin reduced this back to control levels. Addition of a NOSII inhibitor, L-NIL, partially blocked the effect of H3 but not H2 relaxin treatments (Fig. 2); this increase in PI+ve cells was significantly greater than PI+ve cell counts in controls (p < 0.05). In the second experiment, the RXFP3 receptor antagonist, R3 B1-22R, blocked the effect of H3 relaxin but not H2 relaxin treatment (Fig. 3).
Fig 1.

Fig 2.
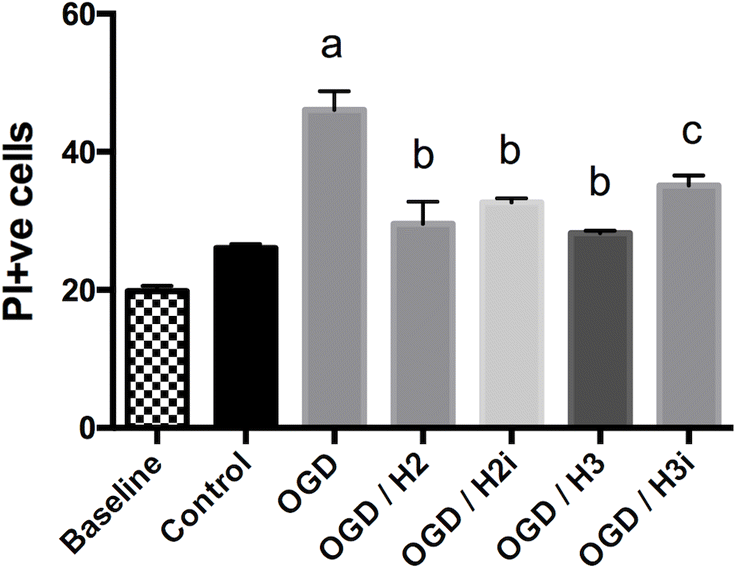
Fig 3.
Discussion
Cultured brain slices exposed to oxygen- and glucose-depleted media for 60 min show an increase in cell damage or death imaged through PI staining. Both relaxin (H2 relaxin) and relaxin-3 (H3 relaxin) treatment reduce cell death in slices and specifically blocking NOS II (or inducible NOS), which partially attenuated the effect of H3 relaxin, suggests that NO is involved, at least in part, in mediating actions of relaxin-3; this supports observations in vivo (Wilson et al. 2006; Bergeron et al. 2015). To determine if neuroprotection is conferred by both relaxin ligands through the binding and activation of RXFP3, deemed to be the relaxin-3 receptor, the RXFP3 antagonist R3 B1-22R was used. RXFP3 antagonism blocked the effect of H3 relaxin, but not H2 relaxin suggesting that additional relaxin receptors may mediate tissue protection from ischemic injury.
In 2002, Hsu et al. (2002) identified an orphan type C GPCR possessing an ectodomain with a leucine-rich repeat and LDL-a module, LGR7, as the cognate receptor for circulating relaxin (H2 relaxin in humans). Later, the SALPR receptor (GPCR135), a GPCR lacking this ectodomain, was determined to be the cognate relaxin-3 receptor (Liu et al. 2003b). After standardizing nomenclature for relaxin-family peptides in 2006 (Bathgate et al. 2006), the revised names of these receptors became relaxin family peptide (RXFP) receptors 1 and 3, respectively. While H2 relaxin and H3 relaxin activate their cognate receptors, in vitro evidence suggests that both ligands can bind RXFP1 and 3 and activate them to different degrees (Liu et al. 2003a, 2003b; Sudo et al. 2003; Halls et al. 2007; van der Westhuizen et al. 2010). Our data suggest that H3 relaxin (relaxin-3) is exerting its actions via RXFP3 as antagonism of this receptor completely blocks its actions on cell survival. RXFP3 antagonism does not affect actions of H2 relaxin that points towards involvement of RXFP1. However, relaxin also binds and activates RXFP2 in cultured cells in vitro (Hsu et al. 2002), causing increases in cAMP. Hsu et al. (2002) found that both RXFP1 and RXFP2 transcripts are expressed in the brain. However, relaxin may not activate RXFP2 in vivo (Bathgate et al. 2013); therefore, the involvement of this receptor is unlikely. Evidence also suggests that H2 and porcine relaxins bind and activate the glucocorticoid receptor in human cell lines (Dschietzig et al. 2004, 2009), implying transcriptomic effects in these cells. However, these effects would be beyond the time frame of the acute effects of relaxins described here; relaxin treatments in anesthetized rats are only effective within 60 min post-MCAO. New specific RXFP1 agonists such as ML-290 and B7-33, and antagonists in development, will facilitate research on the role of this receptor in neuroprotection and perhaps confirm that it mediates neuroprotective actions of H2 relaxin (Hossain et al. 2016; Ng et al. 2019; Praveen et al. 2019). This will permit studies of cellular mechanisms linked to receptor activation.
Another question raised by this study is whether relaxins are affecting survival of neurons and (or) glial cells in cultured brain slices under OGD conditions. Both RXFP1 and RXFP3 gene expression and presence of protein immunostaining has been demonstrated in neocortical neurons of rat and mouse brains, including the level where PI+ve cell counts were made in slices used in this study (Burazin et al. 2005; Gundlach et al. 2009). In terms of glial cells, there appears to be little published data characterizing RXFP receptor expression. Data from Coldren et al. showed RXFP1 immunostaining on astrocytes, but not microglia, in rat brains. Willcox and Summerlee (2014) have shown that both relaxin and relaxin-3 maintain rat primary cortical astrocyte viability during OGD conditions and subsequent work (Bergeron et al. 2015) showed that RXFP1 and RXFP3 are expressed by these cells, with RXFP3 expression much higher than that of RXFP1. In considering future experiments using the organotypic neonatal rat brain slice model to study relaxin ligand or receptor mechanisms that protect neural cells, the presence of receptors on neurons, astrocytes, and other glial cells needs to be verified. Furthermore, glial- and neuronal-specific markers can aid the study of the complex relationship between glial cells and neurons during their response to ischemic injury in the brain.
Blocking NO synthesis partially attenuated the effect of H3 relaxin on cell survival in OGD slices. Previous studies of the neuroprotective actions of relaxins in a rodent model of stroke showed that L-NIO effectively blocked the effect of both H2 and H3 relaxins in reducing infarct size; however, this inhibitor is nonselective for NOS isoforms (Wilson et al. 2006; Bergeron et al. 2015). In the current study using cultured brain slices, L-NIL was used to selectively target NOSII as this form is upregulated in ischemia-reperfusion injury and L-NIL has a 23–28-fold greater inhibitory effect on iNOS vs nNOS, and 49-fold greater effect on iNOS vs eNOS (Moore et al. 1994; Alderton et al. 2001). The 50 μmol/L dose of L-NIL was made up in media with relaxin peptides and applied to slices under OGD conditions in culture. In selecting this dose of L-NIL we considered previous use of this inhibitor in a study using acute brain slice preparations (Brown et al. 1999) and two studies where L-NIL was used to block NOS in activated macrophage in culture (Stenger et al. 1995). It is possible that a higher dose of L-NIL would have been effective at reducing neuroprotective effect of both H2 and H3 relaxins in the current study.
NO has been shown to have both beneficial and deleterious effects in tissues undergoing ischemia–reperfusion (reviewed in Alderton et al. 2001). Apparent contradictory NO effects in ischemic tissues may result from different concentrations, timing, and duration of NO exposure, presence with other free radical species, and the presence or absence of specific signal transduction pathways in different cells. In general, NO tends to be protective at lower levels and toxic at higher levels (reviewed in Brown 2010). In healthy neural tissue, the principle synthases involved in NO production are neuronal NOS (NOSI), localized in neurons and astrocytes in low levels (Downen et al. 1999), and endothelial NOS (NOSIII), localized in neurons, astrocytes, and endothelia (Dinerman et al. 1994; Doyle and Slater 1997; Oka et al. 2004). In ischemia–reperfusion injury, NOSII is produced by microglia and astrocytes in response to pathogens, damage, and hypoxia (Liu et al. 1996; Liu and Neufeld 2000). Along with the inhibitory effects on immune cells and vasodilation described above, NO release may also mediate many protective changes in neural cells such as inhibiting mitochondrial oxygen consumption (increasing available oxygen with vasodilation), protecting mitochondrial integrity, acting as an antioxidant, inhibiting apoptosis in neurons (at low levels; however, NO also induces apoptosis via separate pathways), reducing ER stress, and possibly enhancing DNA repair processes (reviewed in Brown 2010)). Along with NO-induced vasodilatory effects in arterial vessels peripheral to the injury site in vivo, which improve collateral perfusion and limit the expansion of the infarct (Bergeron et al. 2015), relaxins may act via NO-mediated mechanisms in neural cells to suppress components of the cellular stress cascade, which would ultimately lead to cell death. Certainly, the latter might be reflected by our findings that inhibiting inducible NOS partially attenuated relaxin-3 effects in cultured brain slices where circulating blood is absent.
This work confirms the findings from in vivo studies in rodents that relaxin and relaxin-3 protect brain tissue from ischemic damage and that this protection is mediated, at least in part, by activation of RXFP3 and the production of NO. The complex role of relaxin receptors and the diversity of potential signalling pathways requires further study. This research promises to increase our understanding of the role of relaxin ligands and their receptors in protecting tissues throughout the body from damage caused by ischemia and reperfusion injury.
Acknowledgements
The authors would like to thank the staff of the Weston Animal Care Centre, Acadia University, for their expert care of the experimental animals. This work was supported by a Nova Scotia Health Research Foundation (NSHRF) Development/Innovative grant (2011-7440) to BCW, and NSHRF and Acadia studentships to HG and ND.
References
Alderton WK, Cooper CE, and Knowles RG. 2001. Nitric oxide synthases: structure, function and inhibition. Biochemical Journal, 357(Pt 3): 593–615.
Alexiou K, Matschke K, Westphal A, Stangl K, and Dschietzig T. 2010. Relaxin is a candidate drug for lung preservation: relaxin-induced protection of rat lungs from ischemia-reperfusion injury. The Journal of Heart and Lung Transplantation, 29(4): 454–460.
Alexiou K, Wilbring M, Matschke K, and Dschietzig T. 2013. Relaxin protects rat lungs from ischemia-reperfusion injury via inducible NO synthase: role of ERK-1/2, PI3K, and forkhead transcription factor FKHRL1. PloS One, 8(9): e75592.
Banerjee A, Shen PJ, Ma S, Bathgate RA, and Gundlach AL. 2010. Swim stress excitation of nucleus incertus and rapid induction of relaxin-3 expression via CRF1 activation. Neuropharmacology, 58(1): 145–155.
Bani D, Bigazzi M, Masini E, Bani G, and Sacchi TB. 1995. Relaxin depresses platelet aggregation: in vitro studies on isolated human and rabbit platelets. Laboratory Investigation, 73(5): 709–716.
Bani D, Failli P, Bello MG, Thiemermann C, Sacchi TB, Bigazzi M, et al. 1998. Relaxin activates the L-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension, 31(6): 1240–1247.
Bani D, Masini E, Bello MG, Bigazzi M, and Sacchi TB. 1998. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. The American Journal of Pathology, 152(5): 1367–1376.
Bathgate RA, Ivell R, Sanborn BM, Sherwood OD, and Summers RJ. 2006. International Union of Pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacological Reviews, 58(1): 7–31.
Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, and Summers RJ. 2013. Relaxin family peptides and their receptors. Physiological Reviews, 93(1): 405–480.
Bathgate RAD, Samuel CS, Burazin TCD, Layfield S, Claasz AA, Reytomas IGT, et al. 2002. Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene – Novel members of the relaxin peptide family. Journal of Biological Chemistry, 277(2): 1148–1157.
Bathgate RAD, Kocan M, Scott DJ, Hossain MA, Good SV, Yegorov S, et al. 2018. The relaxin receptor as a therapeutic target – perspectives from evolution and drug targeting. Pharmacology & Therapeutics, 187:114–132.
Beiert T, Knappe V, Tiyerili V, Stockigt F, Effelsberg V, Linhart M, et al. 2018. Chronic lower-dose relaxin administration protects from arrhythmia in experimental myocardial infarction due to anti-inflammatory and anti-fibrotic properties. International Journal of Cardiology. 250:21–28.
Bergeron LH, Willcox JM, Alibhai FJ, Connell BJ, Saleh TM, Wilson BC, et al. 2015. Relaxin peptide hormones are protective during the early stages of ischemic stroke in male rats. Endocrinology, 156(2): 638–646.
Boccalini G, Sassoli C, Formigli L, Bani D, and Nistri S. 2015. Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: involvement of the Notch-1 pathway. FASEB Journal, 29(1): 239–249.
Boehnert MU, Armbruster FP, and Hilbig H. 2008. Relaxin as a protective substance in preservation solutions for organ transplantation, as shown in an isolated perfused rat liver model. Transplantation Proceedings, 40(4): 978–980.
Brown GC. 2010. Nitric oxide and neuronal death. Nitric Oxide, 23(3): 153–165.
Brown LA, Key BJ, and Lovick TA. 1999. Bio-imaging of nitric oxide-producing neurones in slices of rat brain using 4,5-diaminofluorescein. Journal of Neuroscience Methods, 92(1–2): 101–110.
Burazin TC, Bathgate RA, Macris M, Layfield S, Gundlach AL, and Tregear GW. 2002. Restricted, but abundant, expression of the novel rat gene-3 (R3) relaxin in the dorsal tegmental region of brain. Journal of Neurochemistry, 82(6): 1553–1557.
Burazin TC, Johnson KJ, Ma S, Bathgate RA, Tregear GW, and Gundlach AL. 2005. Localization of LGR7 (relaxin receptor) mRNA and protein in rat forebrain: correlation with relaxin binding site distribution. Annals of the New York Academy of Sciences, 1041: 205–210.
Olfert ED, Cross BM, and McWilliam AA, (eds.). 1993. Guide to the care and use of experimental animals. CCAC, Vol. 1, p. 195.
Collino M, Rogazzo M, Pini A, Benetti E, Rosa AC, and Chiazza F, et al. 2013a. Acute treatment with relaxin protects the kidney against ischaemia/reperfusion injury. Journal of Cellular and Molecular Medicine, 17(11): 1494–1505.
Collino M, Rogazzoi M, Pini A, Benetti E, Rosa AC, Fantozzi R, et al. 2013b. Acute treatment with relaxin attenuates the injury/dysfunction induced by renal ischemia/reperfusion injury. Italian Journal of Anatomy and Embryology, 118(Suppl 1): 74–76.
Cosen-Binker LI, Binker MG, Cosen R, Negri G, and Tiscornia O. 2006. Relaxin prevents the development of severe acute pancreatitis. World Journal of Gastroenterology, 12(10): 1558–1568.
Dinerman JL, Dawson TM, Schell MJ, Snowman A, and Snyder SH. 1994. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America, 91(10): 4214–4218.
Downen M, Zhao ML, Lee P, Weidenheim KM, Dickson DW, and Lee SC. 1999. Neuronal nitric oxide synthase expression in developing and adult human CNS. Journal of Neuropathology & Experimental Neurology, 58(1): 12–21.
Doyle CA, and Slater P. 1997. Localization of neuronal and endothelial nitric oxide synthase isoforms in human hippocampus. Neuroscience, 76(2): 387–395.
Dschietzig T, Richter C, Bartsch C, Laule M, Armbruster FP, Baumann G, et al. 2001. The pregnancy hormone relaxin is a player in human heart failure. FASEB Journal, 15(12): 2187–2195.
Dschietzig T, Bartsch C, Stangl V, Baumann G, and Stangl K. 2004. Identification of the pregnancy hormone relaxin as glucocorticoid receptor agonist. FASEB Journal, 18(13): 1536–1538.
Dschietzig T, Bartsch C, Baumann G, and Stangl K. 2009. RXFP1-inactive relaxin activates human glucocorticoid receptor: Further investigations into the relaxin-GR pathway. Regulatory Peptides, 154(1–3): 77–84.
Failli P, Nistri S, Quattrone S, Mazzetti L, Bigazzi M, Sacchi TB, et al. 2002. Relaxin up-regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB Journal, 16(2): 252–254.
Gahwiler BH, Capogna M, Debanne D, McKinney RA, and Thompson SM. 1997. Organotypic slice cultures: a technique has come of age. Trends in Neuroscience, 20(10): 471–477.
Gahwiler BH, Thompson SM, and Muller D. 2001. Preparation and maintenance of organotypic slice cultures of CNS tissue. Current Protocols in Neuroscience,
Gundlach AL, Ma S, Sang Q, Shen PJ, Piccenna L, Sedaghat K, et al. 2009. Relaxin family peptides and receptors in mammalian brain. Annals of the New York Academy of Sciences, 1160: 226–235.
Halls ML, van der Westhuizen ET, Bathgate RA, and Summers RJ. 2007. Relaxin family peptide receptors–former orphans reunite with their parent ligands to activate multiple signalling pathways. British Journal of Pharmacology, 150(6): 677–691.
Haugaard-Kedstrom LM, Shabanpoor F, Hossain MA, Clark RJ, Ryan PJ, Craik DJ, et al. 2011. Design, synthesis, and characterization of a single-chain peptide antagonist for the relaxin-3 receptor RXFP3. Journal of the American Chemical Society, 133(13): 4965–4974.
Hossain MA, Kocan M, Yao ST, Royce SG, Nair VB, Siwek C, et al. 2016. A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. Chemical Science, 7(6): 3805–3819.
Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD, et al. 2002. Activation of orphan receptors by the hormone relaxin. Science, 295(5555): 671–674.
Kageyama S, Nakamura K, Fujii T, Ke B, Sosa RA, Reed EF, et al. 2018. Recombinant relaxin protects liver transplants from ischemia damage by hepatocyte glucocorticoid receptor: From bench-to-bedside. Hepatology, 68(1): 258–273.
Liu B, and Neufeld AH. 2000. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia, 30(2): 178–186.
Liu C, Chen J, Sutton S, Roland B, Kuei C, Farmer N, et al. 2003a. Identification of relaxin-3/INSL7 as a ligand for GPCR142. Journal of Biological Chemistry. 278(50): 50765–50770.
Liu C, Eriste E, Sutton S, Chen J, Roland B, Kuei C, et al. 2003b. Identification of relaxin-3/INSL7 as an endogenous ligand for the orphan G-protein-coupled receptor GPCR135. Journal of Biological Chemistry, 278(50): 50754–50764.
Liu J, Zhao ML, Brosnan CF, and Lee SC. 1996. Expression of type II nitric oxide synthase in primary human astrocytes and microglia: role of IL-1beta and IL-1 receptor antagonist. Journal of Immunology, 157(8): 3569–3576.
Martin B, Gabris-Weber BA, Reddy R, Romero G, Chattopadhyay A, and Salama G. 2018. Relaxin reverses inflammatory and immune signals in aged hearts. PloS One, 13(1): e0190935.
Masini E, Bani D, Bigazzi M, Mannaioni PF, and Bani-Sacchi T. 1994. Effects of relaxin on mast cells. In vitro and in vivo studies in rats and guinea pigs. Journal of Clinical Investigation, 94(5): 1974–1980.
Masini E, Bello MGD, Bani D, Bigazzi M, Sacchi TB, and Mannaioni PF. 1995. Relaxin inhibits histamine release from mast cells: involvement of nitric oxide production. Inflammation Research, 44(Suppl 1): S12–S13.
Masini E, Salvemini D, Mugnai L, Bello MG, Bani D, and Mannaioni PF. 1996. The effect of relaxin on myocardial ischaemia-reperfusion injury and histamine release in vitro and in vivo. Inflammation Research, 44(Suppl 1): S27–S28.
Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, and Sacchi TB. 1997. Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology, 138(11): 4713–4720.
Masini E, Nistri S, Vannacci A, Sacchi TB, Novelli A, and Bani D. 2004. Relaxin inhibits the activation of human neutrophils: Involvement of the nitric oxide pathway. Endocrinology, 145(3): 1106–1112.
Masini E, Cuzzocrea S, Mazzon E, Muia C, Vannacci A, Fabrizi F, et al. 2006. Protective effects of relaxin in ischemia/reperfusion-induced intestinal injury due to splanchnic artery occlusion. British Journal of Pharmacology, 148(8): 1124–1132.
Matheson PJ, Walker SK, Maki AC, Shaheen SP, Neal Garrison R, and Downard CD. 2014. Oral relaxin maintains intestinal blood flow in a rat model of NEC. Journal of Pediatric Surgery, 49(6): 961–965; discussion 964–965.
McGowan BM, Stanley SA, Smith KL, White NE, Connolly MM, Thompson EL, et al. 2005. Central relaxin-3 administration causes hyperphagia in male Wistar rats. Endocrinology, 146(8): 3295–3300.
McGowan BM, Stanley SA, Smith KL, Minnion JS, Donovan J, Thompson EL, et al. 2006. Effects of acute and chronic relaxin-3 on food intake and energy expenditure in rats. Regulatory Peptides, 136(1–3): 72–77.
McGowan BM, Stanley SA, White NE, Spangeus A, Patterson M, Thompson EL, et al. 2007. Hypothalamic mapping of orexigenic action and Fos-like immunoreactivity following relaxin-3 administration in male Wistar rats. American Journal of Physiology, 292(3): E913–E919.
McGowan BM, Stanley SA, Donovan J, Thompson EL, Patterson M, Semjonous NM, et al. 2008. Relaxin-3 stimulates the hypothalamic-pituitary-gonadal axis. American Journal of Physiology, 295(2): E278–E286.
Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, and Currie MG. 1994. L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. Journal of Medicinal Chemistry, 37(23): 3886–3888.
Moore XL, Tan SL, Lo CY, Fang L, Su YD, Gao XM, et al. 2007. Relaxin antagonizes hypertrophy and apoptosis in neonatal rat cardiomyocytes. Endocrinology, 148(4): 1582–1589.
Ng HH, Esteban-Lopez M, and Agoulnik AI. 2019. Targeting the relaxin/insulin-like family peptide receptor 1 and 2 with small molecule compounds. Molecular and Cellular Endocrinology, 487: 40–44.
Nistri S, and Bani D. 2005. Relaxin in vascular physiology and pathophysiology: possible implications in ischemic brain disease. Current Neurovascular Research, 2(3): 225–233.
Nistri S, Cinci L, Perna AM, Masini E, and Bani D. 2008. Mast cell inhibition and reduced ventricular arrhythmias in a swine model of acute myocardial infarction upon therapeutic administration of relaxin. Inflammation Research, 57(Suppl. 1): S7–S8.
Oka M, Wada M, Yamamoto A, Itoh Y, and Fujita T. 2004. Functional expression of constitutive nitric oxide synthases regulated by voltage-gated Na+ and Ca2+ channels in cultured human astrocytes. Glia, 46(1):53–62.
Paxinos G, and Watson C. 1998. The rat brain in stereotaxic coordinates. Academic Press, San Diego.
Praveen P, Kocan M, Valkovic A, Bathgate R, and Hossain MA. 2019. Single chain peptide agonists of relaxin receptors. Molecular and Cellular Endocrinology, 487: 34–39.
Smith CM, Shen PJ, Banerjee A, Bonaventure P, Ma S, Bathgate RA, et al. 2010. Distribution of relaxin-3 and RXFP3 within arousal, stress, affective, and cognitive circuits of mouse brain. The Journal of Comparative Neurology, 518(19): 4016–4045.
Smith CM, Ryan PJ, Hosken IT, Ma S, and Gundlach AL. 2011. Relaxin-3 systems in the brain–the first 10 years. Journal of Chemical Neuroanatomy, 42(4): 262–275.
Stenger S, Thuring H, Rollinghoff M, Manning P, and Bogdan C. 1995. L-N6-(1-iminoethyl)-lysine potently inhibits inducible nitric oxide synthase and is superior to NG-monomethyl-arginine in vitro and in vivo. European Journal of Pharmacology, 294(2–3): 703–712.
Strasser U, and Fischer G. 1995. Quantitative measurement of neuronal degeneration in organotypic hippocampal cultures after combined oxygen/glucose deprivation. Journal of Neuroscience Methods, 57(2): 177–186.
Sudo S, Kumagai J, Nishi S, Layfield S, Ferraro T, Bathgate RAD, et al. 2003. H3 relaxin is a specific ligand for LGR7 and activates the receptor by interacting with both the ectodomain and the exoloop 2. Journal of Biological Chemistry, 278(10): 7855–7862.
Sutton SW, Shelton J, Smith C, Williams J, Yun S, Motley T, et al. 2009. Metabolic and neuroendocrine responses to RXFP3 modulation in the central nervous system. Annals of the New York Academy of Sciences, 1160: 242–249.
Valle Raleigh J, Mauro AG, Devarakonda T, Marchetti C, He J, Kim E, et al. 2017. Reperfusion therapy with recombinant human relaxin-2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS-dependent mechanism. Cardiovascular Research, 113(6): 609–619.
Watanabe Y, Miyamoto Y, Matsuda T, and Tanaka M. 2011. Relaxin-3/INSL7 regulates the stress-response system in the rat hypothalamus. Journal of Molecular Neuroscience, 43(2): 169–174.
van der Westhuizen ET, Christopoulos A, Sexton PM, Wade JD, and Summers RJ. 2010. H2 relaxin is a biased ligand relative to H3 relaxin at the relaxin family peptide receptor 3 (RXFP3). Molecular Pharmacology, 77(5): 759–772.
Willcox JM, and Summerlee AJS. 2014. Relaxin protects astrocytes from hypoxia in vitro. PloS One, 9(3): e90864.
Wilson BC, Connell B, and Saleh TM. 2006. Relaxin-induced reduction of infarct size in male rats receiving MCAO is dependent on nitric oxide synthesis and not estrogenic mechanisms. Neuroscience Letters, 393(2–3): 160–164.
Yegorov S, Bogerd J, and Good SV. 2014. The relaxin family peptide receptors and their ligands: new developments and paradigms in the evolution from jawless fish to mammals. General and Comparative Endocrinology, 209: 93–105.
Yoshida T, Kumagai H, Kohsaka T, and Ikegaya N. 2013. Relaxin protects against renal ischemia-reperfusion injury. The American Journal of Physiology - Renal Physiology, 305(8): F1169–F1176.
Yoshida T, Kumagai H, Kohsaka T, and Ikegaya N. 2014. Protective effects of relaxin against cisplatin-induced nephrotoxicity in rats. Nephron Experimental Nephrology, 128(1–2): 9–20.
Information & Authors
Information
Published In

FACETS
Volume 6 • Number 1 • January 2021
Pages: 118 - 130
Editor: Matthew John Hogan
History
Received: 20 April 2020
Accepted: 2 November 2020
Version of record online: 4 February 2021
Copyright
© 2021 DeAdder et al. This work is licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Data Availability Statement
All relevant data are within the paper.
Key Words
Sections
Subjects
Authors
Author Contributions
BCW conceived and designed the study.
All performed the experiments/collected the data.
All analyzed and interpreted the data.
BCW contributed resources.
BCW drafted or revised the manuscript.
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Nicholas P. DeAdder, Hannah J. Gillam, and Brian C. Wilson. 2021. Relaxin peptides reduce cellular damage in cultured brain slices exposed to transient oxygen–glucose deprivation: an effect mediated by nitric oxide. FACETS.
6(): 118-130. https://doi.org/10.1139/facets-2020-0029
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. The Relaxin-3 Receptor, RXFP3, Is a Modulator of Aging-Related Disease