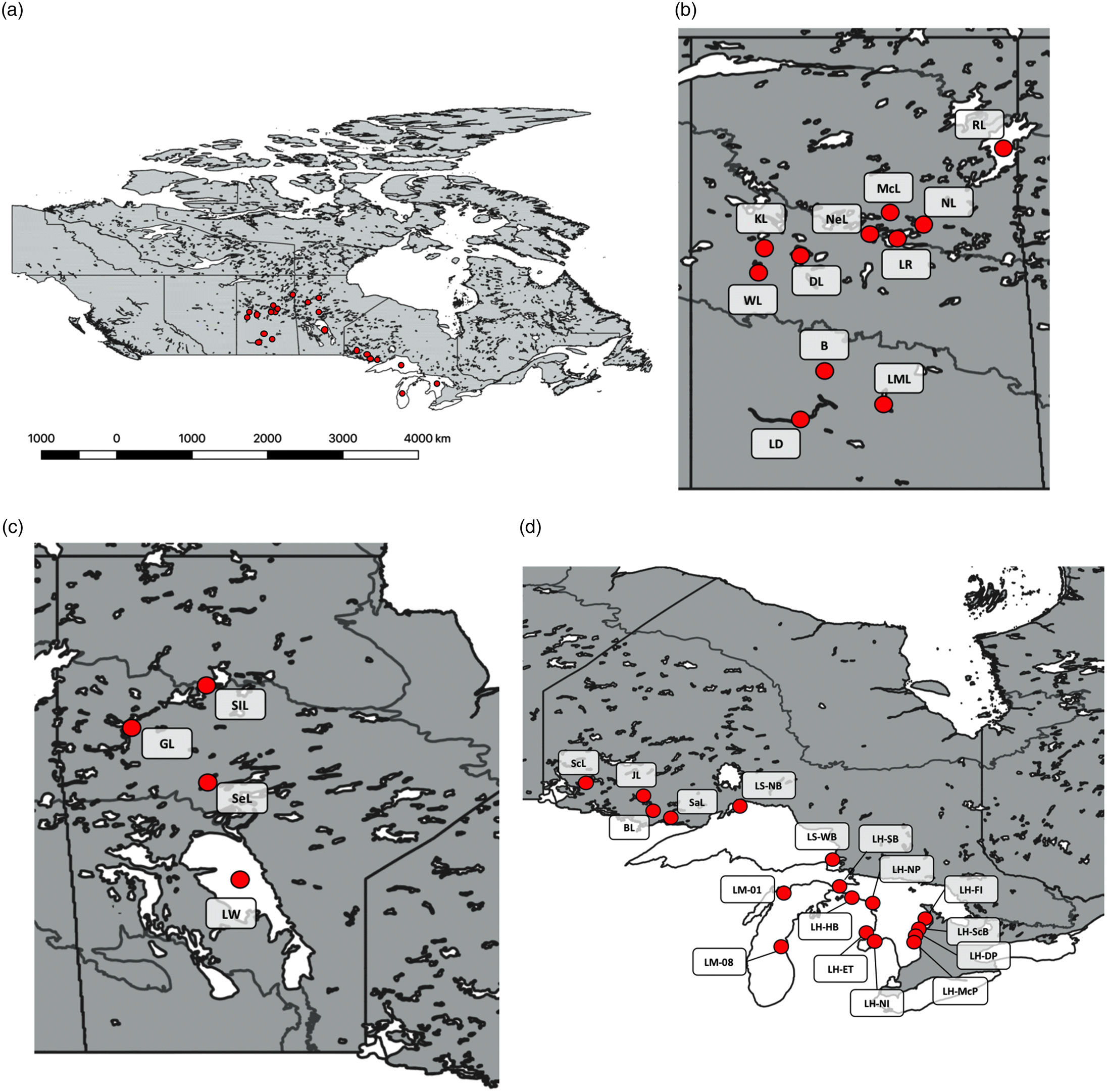
Data quality filtering and genotyping parameters
Following the removal of the Nextera adapters there was a total of 1,911,624,117 reads with an average of 3,740,947 (SD = 2,191,292) reads per individual. After cleaning and trimming in process_radtags there were 3,242,831 (SD = 1,984,622) average reads per individual. The STACKS modules were run to generate a catalog with 3,221,783 markers. This catalog was generated using 124 individuals, each with average numbers of reads. Following sstacks, there was an average of 77,339 (SD = 37,161) matched loci per individual. Filtering and export in the populations module resulted in a total of 3,173 polymorphic SNP loci identified within the dataset.
Following inspection for missing data using
grur, 27 individuals with less than 30% of variable sites genotyped were removed from the analysis, resulting in 481 individuals. Overall, the average percentage of missing data was 18.9% (SD = 14.1%). The IBM plot generated showed that there was no evidence of major biases or clustering of sites based on patterns of missingness for PCo1, explaining 27% of the variance (
Supplementary Material 2, Fig. S1). Some individuals showed divergence based on missingness in other plots along PCo2 and PCo3, but the amount of variance represented was negligible (<2%;
Supplementary Material 2, Fig. S1). Interestingly, minor deviance for Lake Huron samples on PCo3 (
Supplementary Material 2, Fig. S1) corresponded to the different library preparation batches that were used to generate the full dataset. This resulted from a lower average number of reads generated following
process_radtags, with 1,824,308 (SD = 788,510) in the Lake Huron samples compared to 3,728,840 (SD = 2,038,043) average from all other sample sites. The percentage of missing data in the Lake Huron samples was similar to that of the samples overall (20.0%, SD = 16.0% overall; 15.9%, SD = 5.4% in Lake Huron). Although there were significant differences in the level of missing data across lakes, Lake Huron was not an outlier and had similar values to many other lakes in the study (Kruskal–Wallis-ANOVA, df = 21, 459,
P < 0.001;
Supplementary Material 2, Fig. S2a).
Using the filter_hwe() function in grur, 34 markers were identified that did not conform to HWE in at least 11 of the 22 populations (P ≤ 0.01). These markers were removed from subsequent analyses that assume HWE, specifically from the FST and maximum likelihood analysis.
Population differentiation analyses
Most populations sampled had observed heterozygosities similar to expected values, with an average observed heterozygosity of 0.266 (SD = 0.044;
Table 2). Populations in each sub-drainage had similar average heterozygosities with values of 0.285 (SD = 0.032) in the Great Lakes, 0.261 (SD = 0.036) in Winnipeg/Nelson, 0.253 (SD = 0.048) in Churchill, and 0.300 (SD = 0.051) in South Saskatchewan/Qu’appelle. Average G
IS across populations from all sites was 0.039 (SD = 0.117;
Table 2). Average G
IS values were negligible from each sub-drainage with values of 0.041 (SD = 0.117) in the Great Lakes, 0.003 (SD = 0.108) in Winnipeg/Nelson, 0.095 (SD = 0.092) in Churchill, and -0.082 (SD = 0.146) in South Saskatchewan/Qu’appelle (
Table 2). The average nucleotide diversity (π) across all sampled populations was 0.000883 (SD = 0.000134;
Table 2). The average nucleotide diversity was similar in the Winnipeg/Nelson, Churchill and South Saskatchewan/Qu’appelle sub-drainages with values of 0.000848 (SD = 0.000167), 0.000876 (SD = 0.000145), and 0.000876 (SD = 0.000082), respectively, while the Great Lakes had a higher average of 0.000984 (SD = 0.000004), which may be the result of a larger number of samples from these lakes (
Table 2).
IBD was tested across all samples because sites farther apart geographically often have populations that are more different genetically based on distance. The mantel test including all samples resulted in an insignificant relationship between geographic and genetic distance with an R value of −0.158 (
P = 0.939). The dbMEM analysis resulted in 19 significant spatial variables as determined using forward selection, indicating that spatial factors are important factor for the partitioning of genetic variance (
Supplementary Material 2, Table S2).
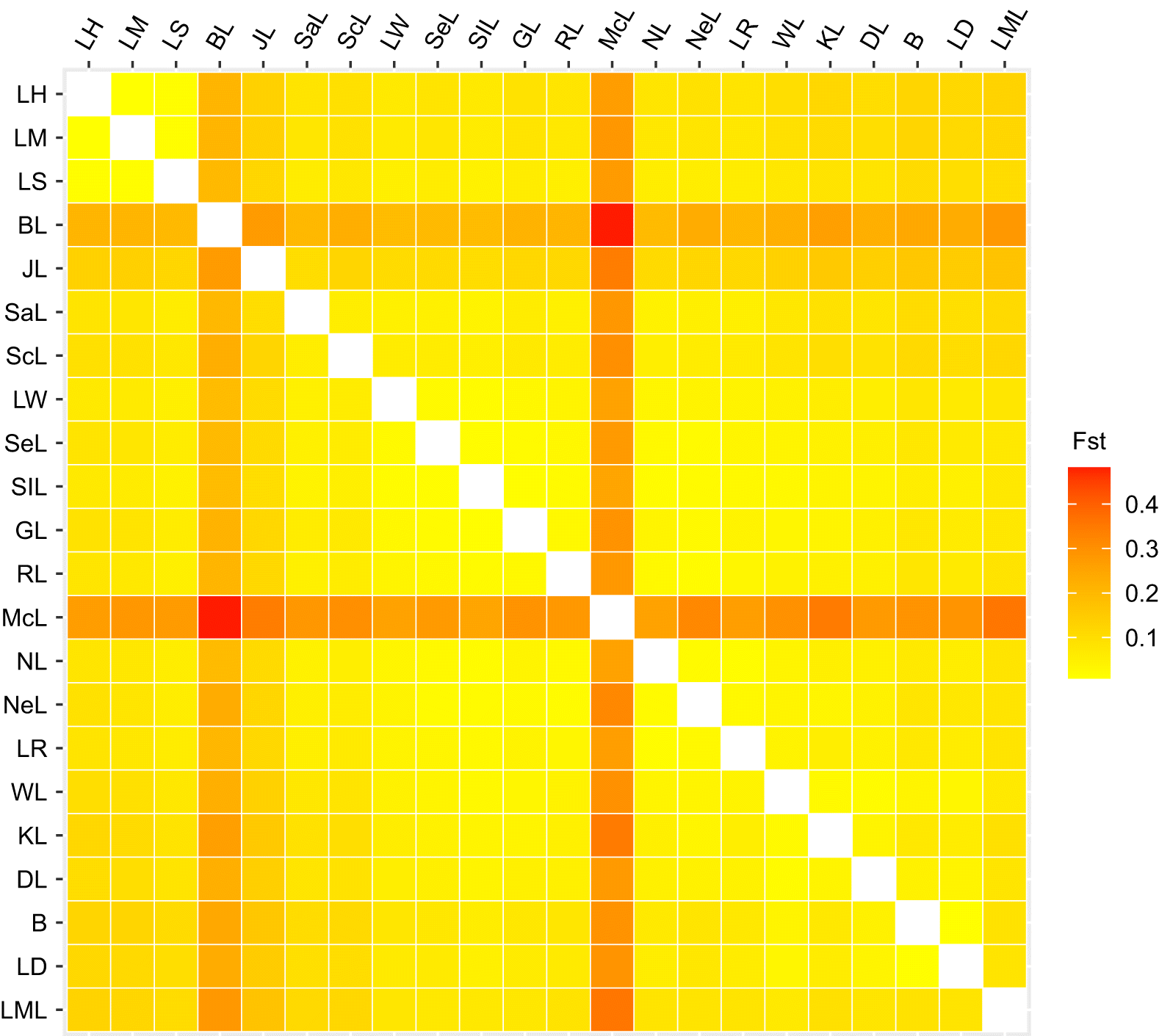
Three population differentiation analyses were used to examine subdivision among populations across the entire sampled range. First, we used GENODIVE to determine pairwise F
ST values between populations in all lakes (
Supplementary Material 2, Table S3). Overall, comparisons among most lakes sampled from different sub-drainages resulted in significant F
ST values. Within each sub-drainage, the average F
ST values were 0.011 (SD = 0.003), 0.122 (SD = 0.077), 0.085 (SD = 0.104), and 0.060 (SD = 0.043) among populations in the Great Lakes, Winnipeg/Nelson, Churchill, and South Saskatchewan/Qu’appelle sub-drainages, respectively (
Fig. 2;
Supplementary Material 2, Table S3). Across sub-drainages, Winnipeg/Nelson and South Saskatchewan/Qu’appelle had the largest average F
ST values of 0.128 (SD = 0.066), followed by 0.114 (SD = 0.010) between Great Lakes and South Saskatchewan/Qu’appelle, 0.114 (SD = 0.097) between Winnipeg/Nelson and Churchill, 0.105 (SD = 0.052) between Great Lakes and Winnipeg/Nelson, 0.098 (SD = 0.062) between Great Lakes and Churchill, and 0.091 (SD = 0.077) between Churchill and South Saskatchewan/Qu’appelle (
Fig. 2;
Table S3). Fish in Mackay Lake in Saskatchewan showed the most differentiation from all other sites, with an average F
ST value of 0.299 (SD = 0.051), followed by Burt Lake with an average F
ST value of 0.232 (SD = 0.063). Some lakes in the Churchill River system (KL, NeL, and NL; see Table 1 for the list of site abbreviations) resulted in some comparisons that were not significant after Bonferroni correction (
Supplementary Material 2, Table S3). Also, some comparisons including BL and LML had F
ST values that were not significant following Bonferroni correction, but this could be due to sample size as the comparisons still resulted in high F
ST values (
Supplementary Material 2, Table S3).
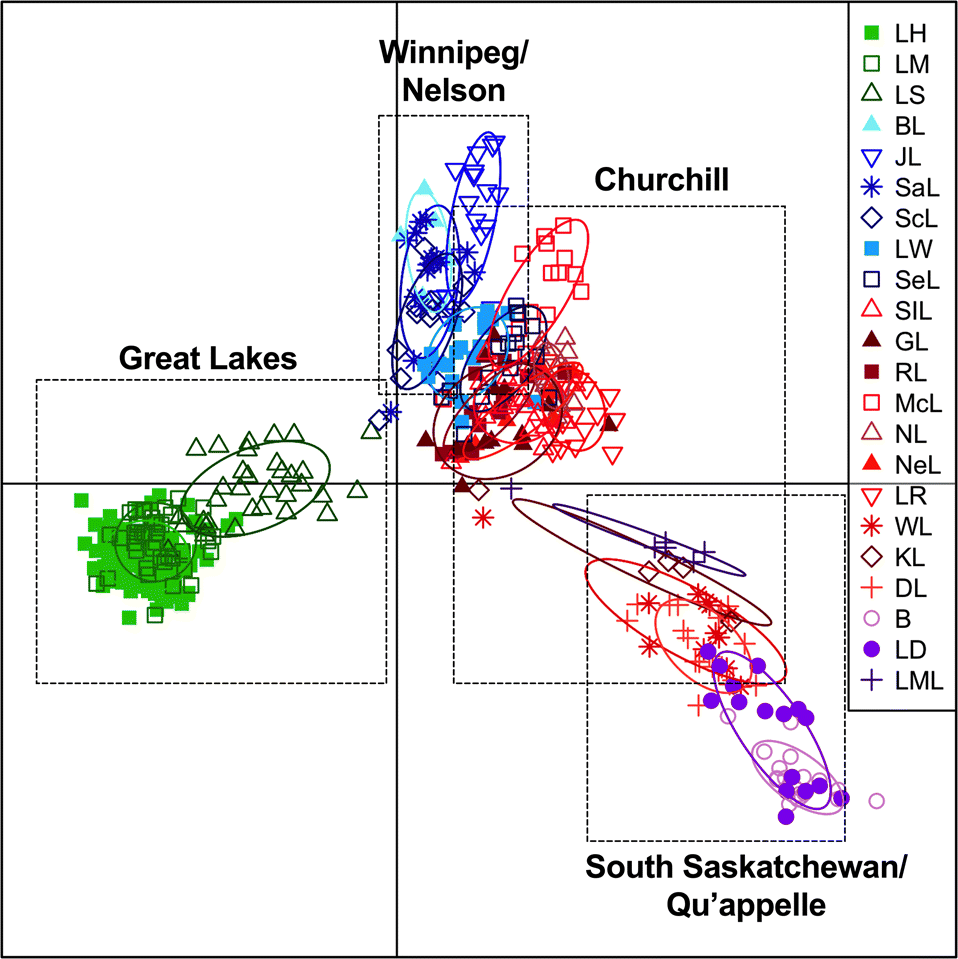
We ran DAPC with all sampled populations using 15 principal components as determined using the
optim_a_score() function. Whitefish populations were significantly differentiated at different spatial scales both within and between provinces. This analysis broadly differentiated populations from the different lakes by sub-drainage and geographic distance along the first axis, explaining 39.2% of the variation. Specifically, along the first axis the Great Lakes were separated from the rest of the sites (
Fig. 3). Lakes in the upper Churchill River (KL, DL and WL), South Saskatchewan River (B and LD), and Qu’appelle River (LML) were also differentiated along the first axis (
Fig. 3). The second axis explained 15.3% of the variation and showed a gradient of variation by distance based on sub-drainage, with lakes in the Winnipeg and Nelson River sub-drainages differentiating from lakes in the Churchill River sub-drainage and South Saskatchewan and Qu’appelle sub-drainages (
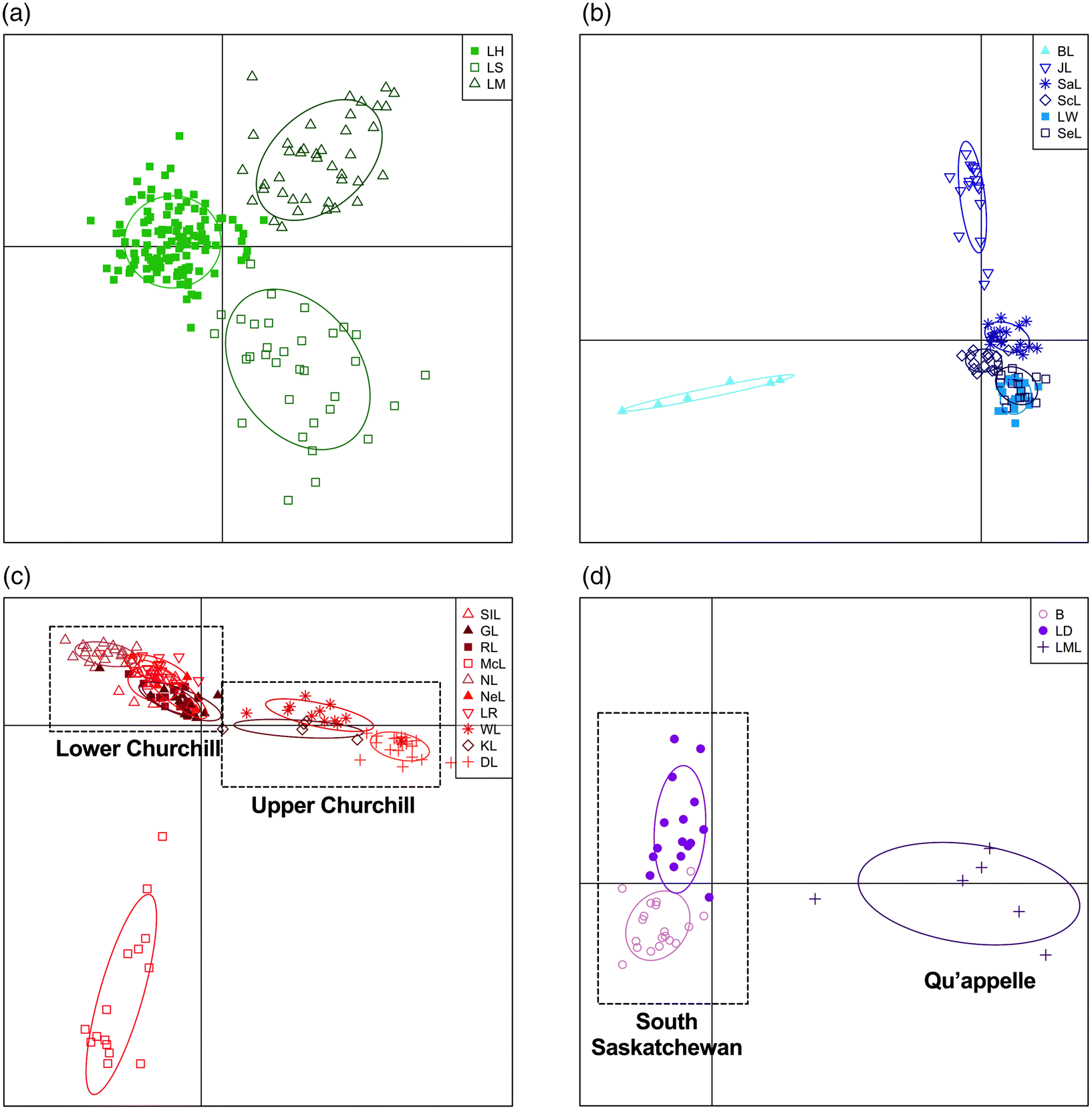
Fig. 3). The average assignment proportion of each individual in the broad analysis was high with a value of 0.892. Differentiation was also detected on sub-drainage scale, running the DAPC using 7 principal components in the Great Lakes, 6 in Lake Winnipeg/Nelson River, 8 in the Churchill River, and 3 in the South Saskatchewan and Qu’appelle sub-drainages (
Fig. 4a–d). The first two axes in the Great Lakes analysis explain 52.3% and 47.7%, respectively, and separated each of the Great Lakes (
Fig. 4a). The DAPC analysis of the Lake Winnipeg and Nelson River sub-drainages explained 29.7% along the first axis and separated BL and explained 27.5% along the second axis and differentiated JL (
Fig. 4b). The first axis of the Churchill River sub-drainage explained 39.9% and differentiated upper and lower Churchill River samples, while the second axis explained 37.9% and separated McL (
Fig. 4c). The Qu’appelle River and South Saskatchewan River samples were differentiated along the first axis of the DAPC analysis, explaining 85.2% and B and LD in the South Saskacthewan River sub-drainage were separated along the second axis, with 14.8% of the of the variation (
Fig. 4d). When run independently, each sub-drainage had high assignment proportions with 0.980 in the Great Lakes, 1.000 in Lake Winnipeg/Nelson River, 0.864 in the Churchill River and 0.902 in South Saskatchewan/Qu’appelle.
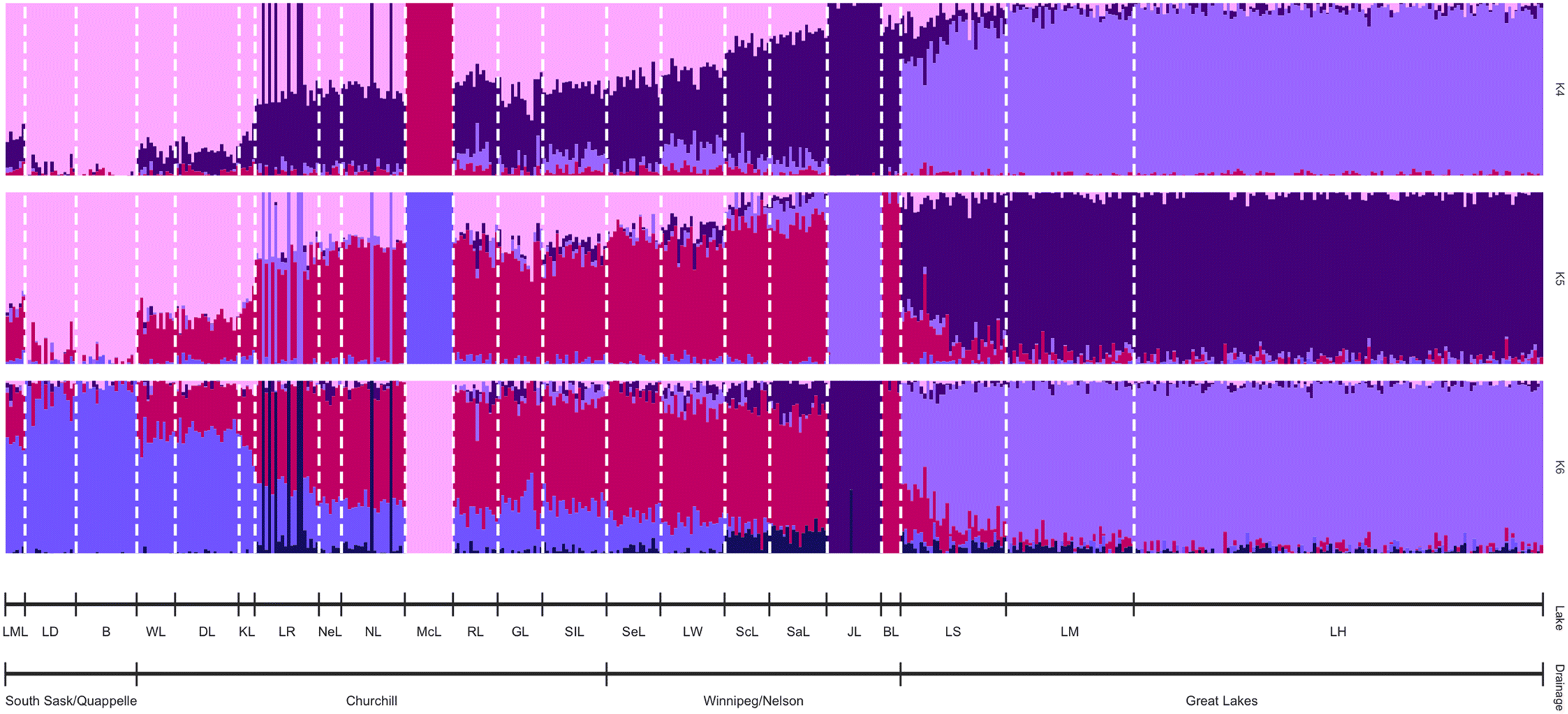
The optimal number of clusters (K) was determined using the cross-validation (CV) technique in ADMIXTURE. The three K values with the lowest CV were retained with K = 4 having a CV value of 0.512, K = 5 with a value of 0.511, and K = 6 with a value of 0.510. Importantly, all three K values detected a gradient of differentiation both on a broad and sub-drainage scale (
Fig. 5). Each K value had high ancestry coefficients of 0.799 (SD = 0.200) for K = 4, 0.825 (SD = 0.147) for K = 5, and 0.808 (SD = 0.150) for K = 6 (
Fig. 5). Further, within sub-drainages we detected differentiation based on hydrological connectivity and geographic distance between sampled populations. This differentiation reflected the results of the DAPC analysis with BL, JL, and McL showing differentiation from other lakes within their sub-drainage (
Fig. 5). Within the Churchill sub-drainage there was a distinct gradient based on connectivity with populations from the upper Churchill River (KL, WL, and DL) slightly differentiating from those in the lower Churchill River (SIL, GL, RL, NL, NeL, and LR;
Fig. 5). Similar to previous analyses, fish from McL strongly differentiated in all three K values with average ancestry coefficients of 0.999 (SD = 1.81 × 10
−6), 0.999 (SD = 1.49 × 10
−16), and 0.999 (SD = 0.00) for the K = 4, K = 5, and K = 6 data, respectively (
Fig. 4). The Great Lakes populations sampled were differentiated from those at the other sites in Ontario with average ancestry coefficients of 0.922 (SD = 0.086) in K = 4, 0.905 (SD = 0.089) in K = 5, and 0.894 (SD = 0.090) in K = 6 (
Fig. 5).